Tradename: Semavic®
International non-proprietary name (INN): semaglutide
Dosage form: solution for subcutaneous injection
Composition: 1 mL of the solution contains 1.34 mg of semaglutide as the active ingredient, as well as excipients: 1.42 mg of disodium hydrogen phosphate dihydrate, 14.0 mg of propylene glycol, 5.5 mg of phenol, diluted 10 % hydrochloric acid and/or 10 % sodium hydroxide solution for pH adjustment to 7.4, water for injection up to 1 mL.
Appearance: clear, colourless or almost colourless solution.
Pharmacotherapeutic group of the medicinal product: hypoglycaemic agent, an analogue of glucagon-like peptide-1 (GLP-1).
ATC code: А10В106.
Pharmacological properties
Semaglutide is a chemically synthesised agonist of glucagon-like peptide-1 receptors (GLP-1R). Semaglutide is a GLP-1 analogue that has 94 % homology with human GLP-1. Semaglutide acts as a GLP-1R agonist that selectively binds to and activates GLP-1R. GLP-1R is a target for native GLP-1. GLP-1 is a physiological hormone that has multiple effects on the regulation of glucose concentration, appetite, and the cardiovascular system (CVS). The effects on glucose concentration and appetite are specifically mediated by GLP-1R located in the pancreas and brain. Pharmacological concentrations of semaglutide reduce blood glucose levels and body weight through a combination of effects described below. GLP-1Rs are also presented in specific areas of heart, vessels, immune system and kidneys, where their activation may have cardiovascular (CV) and microcirculatory effects. Unlike native GLP-1, prolonged elimination half-life of semaglutide (about 1 week) allows it to be used subcutaneously (s.c.) once a week. The main mechanism of the prolonged action of semaglutide is its binding to albumin, resulting in reduced renal elimination and protection from metabolic breakdown. Moreover, semaglutide is resistant to degradation by enzyme dipeptidyl peptidase-4. Semaglutide reduces blood glucose levels through glucose-dependent stimulation of insulin secretion and glucose-dependent suppression of glucagon secretion. Hence, with the increase of blood glucose concentration, insulin secretion is stimulated and glucagon section is inhibited. The glucose-lowering mechanism also involves a slight delay in early postprandial stomach emptying. In hypoglycaemia, semaglutide reduces insulin secretion but does not reduce glucagon secretion. Semaglutide helps reduce overall body weight and the weight of subcutaneous fat tissue while reducing energy consumption. This mechanism occurs via the reduction of appetite, enhancing satiation signals, and better control of food intake cravings. Insulin resistance also decreases, perhaps, due to weight loss. In addition, semaglutide lowers a preference for high-fat foods. Animal studies have shown that semaglutide is absorbed by specific areas of the brain, enhances key saturation signals, and attenuates key hunger signals. By acting on isolated areas in brain tissues, semaglutide activates satiety-related neurons and inhibits hunger-related neurons. Semaglutide had a beneficial effect on plasma lipids, lowered systolic blood pressure (BP) and reduced inflammation in clinical studies (CS). In animal studies, semaglutide attenuates the development of atherosclerosis by preventing aortic plaque progression and reducing inflammation in the plaque.
Pharmacodynamics
All pharmacodynamic evaluations were performed after 12 weeks of treatment (including dose escalation) at steady state with semaglutide 1 mg once weekly.
Fasting and postprandial blood glucose levels
Semaglutide decreases fasting and postprandial glucose concentrations. Compared to placebo, therapy with semaglutide 1 mg in patients with type 2 diabetes mellitus (T2D) resulted in a decrease in glucose concentrations in terms of absolute change from baseline (mmol/L) and relative decrease compared to placebo (%) in terms of: fasting glucose concentrations (1.6 mmol/L; 22 %); glucose concentrations 2 hours after eating (4.1 mmol/L; 37 %); mean daily glucose concentration (1.7 mmol/L; 22 %); and postprandial peak glucose concentrations for 3 meals (0.6–1.1 mmol/L). Semaglutide lowered fasting glucose after the first dose.
Pancreatic beta cell function and insulin secretion
Semaglutide decreases fasting and postprandial glucose concentrations. Compared to placebo, therapy with semaglutide 1 mg in patients with type 2 diabetes mellitus (T2D) resulted in a decrease in glucose concentrations in terms of absolute change from baseline (mmol/L) and relative decrease compared to placebo (%) in terms of: fasting glucose concentrations (1.6 mmol/L; 22 %); glucose concentrations 2 hours after eating (4.1 mmol/L; 37 %); mean daily glucose concentration (1.7 mmol/L; 22 %); and postprandial peak glucose concentrations for 3 meals (0.6–1.1 mmol/L). Semaglutide lowered fasting glucose after the first dose.
Glucagon secretion
Semaglutide decreases fasting and postprandial glucagon concentrations. In patients with T2D, semaglutide induces a relative decrease in glucagon concentrations compared to placebo: fasting glucagon concentrations (8–21 %), postprandial glucagon secretion (14–15 %), and mean daily glucagon concentrations (12 %).
Glucose-dependent insulin and glucagon secretion
Semaglutide lowered high blood glucose concentrations by stimulating insulin secretion and lowering glucagon secretion in a glucose-dependent manner. With semaglutide, the insulin secretion rate in T2D patients was comparable to that of healthy volunteers. During induced hypoglycaemia, semaglutide compared to placebo did not alter the counter regulatory responses of increased glucagon and did not impair the decrease of C-peptide in patients with type 2 diabetes.
Gastric emptying
Semaglutide caused a minor delay of early postprandial gastric emptying, thereby reducing the rate at which glucose appears in the circulation postprandially
Body weight and constitution
Greater weight loss was observed with semaglutide compared to other studied comparators (placebo, sitagliptin, extended release exenatide, dulaglutide and insulin glargine) (see section “Clinical efficacy and safety”). The body weight loss with semaglutide is mostly due to the fat tissue loss 3 times greater than the muscle tissue loss.
Appetite, energy intake and food choice
Semaglutide compared to placebo lowered the energy intake of three consecutive ad libitum meals by 18–35 %. This was supported by a semaglutide-induced suppression of appetite in the fasting state as well as postprandially, improved control of eating, less food cravings and a relative lower preference for high fat food.
Fasting and postprandial lipids
Compared to placebo, semaglutide decreased fasting triglyceride and very low-density lipoprotein (VLDL) cholesterol concentrations by 12 % and 21 %, respectively. In response to a high-fat meal, increased postprandial triglyceride and VLDL cholesterol concentrations were lowered by more than 40 %.
Cardiac electrophysiology (CEP)
The effect of semaglutide on cardiac repolarisation was tested in the CEP study. Semaglutide did not prolong corrected QT intervals at dose levels higher than therapeutic doses (up to 1.5 mg at steady state).
Pharmacokinetics
The medicinal product is suitable for once-weekly injection as the elimination half-life of semaglutide is approximately 1 week.
Absorption
Maximum plasma concentration (Cmax) was reached 1 to 3 days post dose. The steady state concentration (AUCt/24) was achieved after 4–5 weeks of once-weekly injection. In patients with type 2 diabetes, the mean steady state concentrations following s.c. injection of semaglutide 0.5 mg and 1 mg were approximately 16 nmol/L and 30 nmol/L, respectively. Semaglutide exposure increased in a dose proportional manner for doses of 0.5 mg and 1 mg. Similar exposure was achieved with s.c. injection of semaglutide in the abdomen, thigh, or upper arm. The absolute bioavailability of semaglutide after s.c. injection reached 89 %.
Distribution
The mean volume of distribution of semaglutide in tissues following s.c. injection in patients with type 2 diabetes was approximately 12.5 L. Semaglutide was significantly bound to plasma albumin (>99 %).
Biotransformation
Semaglutide is metabolised via proteolytic cleavage of the peptide backbones and sequential beta-oxidation of the fatty acid side chain.
Elimination
Gastrointestinal tract and kidneys were the primary excretion routes of semaglutide. 2/3 of semaglutide dose are excreted in urine and 1/3 in faeces. Approximately 3 % of the dose are excreted as intact semaglutide via kidneys. Clearance of semaglutide in patients with type 2 diabetes was approximately 0.05 L/h. With an elimination half-life of approximately 1 week, semaglutide will be present in the circulation for about 5 weeks after the last dose.
Special populations
No dose adjustment for semaglutide is required depending on age, gender, race and ethnicity, body weight, presence of renal or hepatic impairment.
Age
Age had no effect on the pharmacokinetics of semaglutide based on data from Phase 3a studies including patients of 20–86 years of age.
Gender
Gender had no effect on the pharmacokinetics of semaglutide.
Race
The race (White, Black or Afro-American, Asian) did not affect the pharmacokinetics of semaglutide.
Ethnicity
Ethnicity (Latino) had no effect on the pharmacokinetics of semaglutide.
Body weight
Body weight has an effect on the exposure of semaglutide. Higher body weight results in lower exposure. Semaglutide doses of 0.5 mg and 1 mg provide adequate systemic exposure over a body weight range of 40–198 kg.
Renal impairment
Renal impairment had no clinically significant effect on the semaglutide pharmacokinetics. This was shown in a study with a single dose of 0.5 mg semaglutide in patients with different severity renal impairment (mild, moderate, severe, or dialysis patients) versus patients with normal renal function. This was also shown for subjects with type 2 diabetes and renal insufficiency based on data from phase 3 studies, though the practical use in patients with end-stage renal disease was limited.
Hepatic impairment
Hepatic impairment had no effect on the semaglutide exposure. The pharmacokinetics of semaglutide was evaluated in a study involving patients with different severity of hepatic impairment (mild, moderate, severe) versus patients with normal hepatic function exposed to a single 0.5 mg dose of semaglutide.
Paediatric population
Semaglutide has not been studied in children and adolescents under 18 years.
Therapeutic indications
Semavic®, in addition to diet and exercise, is indicated for adult patients with type 2 diabetes mellitus to improve glycaemic control as:
Semavic® is indicated to reduce the risk of major cardiovascular events* in patients with type 2 diabetes mellitus and high cardiovascular risk as an add-on to a standard cardiovascular therapy (based on the time of onset of the first major cardiovascular event (see section “Pharmacological properties, subsection assessment of effect on CVS”)).
*major cardiovascular events include cardiovascular death, non-fatal myocardial infarction, and non-fatal stroke.
Contraindications
Semavic® is contraindicated in the following populations and for the following conditions/diseases due to the missing efficacy and safety data or limited experience:
Precautions
Semavic® is recommended to be used with caution in patients with renal impairment and in patients with a history of pancreatitis (see section “Special warnings”).
Use during pregnancy and breastfeeding
Pregnancy
Animal studies demonstrated the reproductive toxicity of the medicinal product (see section “Non-clinical safety data”). The data on the use of semaglutide in pregnant women is limited. Semaglutide is not contraindicated during pregnancy. Women of childbearing potential are recommended to use methods of contraception during therapy with semaglutide. If a patient wishes to become pregnant, or pregnancy occurs, semaglutide should be discontinued. Semaglutide should be discontinued at least 2 months before a planned pregnancy due to the long half-life (see section “Pharmacokinetics”).
Breastfeeding
Semaglutide was excreted in the milk of lactating rats. A risk to a breastfed baby cannot be excluded. Semaglutide should not be used for the duration of breastfeeding.
Posology and method of administration
Method of administration
Use Semavic® once weekly, any time of the day, regardless of meals. Inject Semavic® subcutaneously (s.c.) in the abdomen, thigh, or upper arm. Injection site rotation does not call for dose adjustment. Do not inject Semavic® intravenously or intramuscularly. Details of the method of administration see in the section “Instruction for use”. The day of weekly dosing can be changed as appropriate, as long as the interval between two doses is at least 3 days (>72 hours).
Posology
The starting dose of Semavic® is 0.25 mg once weekly. After 4 weeks of administration, the dose should be increased to 0.5 mg once weekly. To further improve glycaemic control, after at least 4 weeks of 0.5 mg once weekly, the dose can be increased to 1 mg once weekly. Semavic® 0.25 mg is not a therapeutic dose. Semavic® can be used as monotherapy or in combination with one or more antidiabetic products (see section “Clinical efficacy and safety”). When Semavic® is added to the existing therapy with metformin and/or thiazolidinedione, the same doses of metformin and/or thiazolidinedione can be continued. When Semavic® is added to the existing therapy with sulfonylurea derivatives or insulin, consider the reduction of the sulfonylurea derivative or insulin dose to minimize the risk of hypoglycaemia (see section “Special warnings”). With the Semavic® therapy, self-monitoring of blood glucose is not required. When Semavic® is co-administered with a sulfonylurea derivative or insulin, self-monitoring of blood glucose may be required to adjust the dose of the sulfonylurea derivative or insulin.
Missed dose
If the dose of Semavic® has been missed, take it as soon as possible, within 5 days of the scheduled dosing. If the dose has been missed for more than 5 days, do not take the missed dose. Take the next dose of Semavic® on the scheduled day. In each case, the patient can resume their usual weekly dosing schedule.
Use of the medicinal product in special populations
Paediatric population
Semavic® is contraindicated in children under 18 years of age due to missing efficacy and safety data.
Elderly (≥65 years old)
No dose adjustment is required based on age. The clinical experience of the use of semaglutide in patients 75 years of age or older is limited.
Patients with renal impairment
No dose adjustment is required for patients with renal impairment. The clinical experience with Semavic® in patients with end-stage renal disease is lacking. Semavic® is contraindicated in such patients.
Patients with hepatic impairment
No dose adjustment is required in patients with hepatic impairment (see section “Pharmacokinetics”). The clinical experience of the use of semaglutide in patients with severe hepatic impairment is limited. Semavic® is contraindicated for such patients.
Side effect
The most frequently reported adverse reactions (AR) in the clinical study were gastrointestinal disorders, including nausea, diarrhoea and vomiting. In general, these reactions were mild or moderate in severity and of short duration. Adverse reactions that may occur with the use of Semavic® are classified by system organ classes as per the MedDRA, with indicating their frequencies in accordance with the WHO recommendations: very common (≥1/10), common (≥1/100, but <1 /10), uncommon (≥1/1000, but <1/100), rare (≥1/10,000, but <1/1000), very rare (<1/10,000), unknown (cannot be assessed on the basis of available data). Within each frequency grouping, adverse reactions are presented in order of decreasing seriousness.The most frequently reported adverse reactions (AR) in the clinical study were gastrointestinal disorders, including nausea, diarrhoea and vomiting. In general, these reactions were mild or moderate in severity and of short duration. Adverse reactions that may occur with the use of Semavic® are classified by system organ classes as per the MedDRA, with indicating their frequencies in accordance with the WHO recommendations: very common (≥1/10), common (≥1/100, but <1/10), uncommon (≥1/1000, but <1/100), rare (≥1/10,000, but <1/1000), very rare (<1/10,000), unknown (cannot be assessed on the basis of available data). Within each frequency grouping, adverse reactions are presented in order of decreasing seriousness.
Table 1. Adverse reactions from Phase 3 clinical study
| Organ system (MedDRA) | Very common | Common | Uncommon | Rare |
|---|---|---|---|---|
| Immune system disorders | Anaphylactic reactions | |||
| Metabolism and nutrition disorders | Hypoglycaemia⁽ᵃ⁾ when co-administrated with insulin or sulphonylurea | Hypoglycaemia⁽ᵃ⁾ when used with OHMPs / Decreased appetite | ||
| Nervous system disorders | Dizziness | Dysgeusia | ||
| Eye disorders | Diabetic retinopathy complications⁽ᵇ⁾ | |||
| Cardiac disorders | Increased heart rate (HR) | |||
| GI disorders | Nausea, diarrhoea | Vomiting, abdominal pain, bloating, constipation, dyspepsia, gastritis, gastroesophageal reflux disease, belching, flatulence | ||
| Hepatobiliary disorders | Cholelithiasis | |||
| General disorders and administration site conditions | Fatigue | Injection site reactions | ||
| Investigations | Increased serum lipase, increased serum amilase, weight loss |
⁽ᵃ⁾ Hypoglycaemia defined as severe (requiring assistance from another person) or symptomatic, in combination with a plasma glucose concentration < 3.1 mmol/L
⁽ᵇ⁾Diabetic retinopathy complications mean a combined need for retinal photocoagulation, need for treatment with intravitreal agents, vitreous haemorrhage, and diabetes-related blindness.
The frequency is based on cardiovascular (CV) outcomes.
Description of selected adverse reactions
Hypoglycaemia
No episodes of severe hypoglycaemia were observed during monotherapy with Semavic®. Severe hypoglycaemia was primarily observed when Semavic® was administered in combination with sulphonylurea derivative or insulin. Several episodes of severe hypoglycaemia were observed when Semavic® was administered in combination with other medicinal products, except for sulphonylureas, OHMPs.
Gastrointestinal ARs
During therapy with Semavic® at doses of 0.5 mg and 1 mg, patients had nausea, diarrhoea, and vomiting. Most reactions were mild to moderate and short-term. The ARs caused withdrawal from the clinical study for 3.9 % and 5.9 % patients, respectively. The ARs were most often reported in the first months of therapy.
Diabetic retinopathy complications
In a 2-year clinical study involving patients with type 2 diabetes and high CV risk, long-term diabetes and inadequate glycaemic control, confirmed cases of diabetic retinopathy complications were observed in more patients in the Semavic® group (3.0 %) vs. placebo (1.8 %). Patients with a history of diabetic retinopathy at the clinical study baseline had a higher absolute risk of complications. Patients with no documented history of diabetic retinopathy had the similar number of events with administration of Semavic® and placebo. In up to 1-year CSs, the incidence of ARs associated with diabetic retinopathy was similar in the Semavic® and comparator groups.
Treatment discontinuation due to an adverse reaction
The incidence of discontinuation of treatment due to NR in 1 mg Semavic® group was 8.7 %. The most frequent ARs leading to discontinuation from treatment were gastrointestinal disorders.
Injection site reactions
Injection site reactions (e. g. rash, erythema at the injection site) were reported in 0.6 % and 0.5 % of patients treated with semaglutide 0.5 and 1 mg, respectively. These reactions were mild, as a rule.
Immunogenicity
Due to the potential immunogenic properties of protein and peptide medicinal products, patients may develop antibodies to semaglutide after therapy with Semavic®. At the end of the study, the share of patients with developed antibodies to semaglutide was low (1–2 %), and none developed semaglutide-neutralising antibodies or semaglutide ADAs with endogenous GLP-1 neutralising effect.
Debido a las posibles propiedades inmunogénicas de los medicamentos proteicos y peptídicos, los pacientes pueden desarrollar anticuerpos contra la semaglutida después del tratamiento con Semavic®. Al final del EC, la proporción de pacientes que presentaban anticuerpos contra la semaglutida en cualquier punto temporal era baja (1–2 %), y no se encontraron pacientes con anticuerpos neutralizantes contra la semaglutida o anticuerpos con un efecto neutralizante sobre el GLP-1 endógeno.
Overdose
Symptoms
Overdoses of up to 4 mg in a single dose, and up to 4 mg in a week have been reported in clinical studies. The most common AR reported was nausea. All patients recovered without complications.
Treatment
There is no specific antidote for overdose with Semavic®. In case of overdose, appropriate symptomatic therapy should be initiated. A prolonged follow-up and treatment period for these symptoms may be necessary, taking into account the long half-life of semaglutide (approximately 1 week).
Interaction with other medicinal products
Pharmacodynamic interaction
In vitro studies of semaglutide have shown a very low potential for inhibition or induction of cytochrome P450 (CYP) enzymes and inhibition of medicinal product transporters. The delay of gastric emptying with the use of semaglutide may influence the absorption of concomitant oral medications.
Semaglutide should be used with caution in patients receiving oral medications that require rapid absorption from the gastrointestinal tract.
Paracetamol
In assessment of the paracetamol pharmacokinetics during a standard meal test, semaglutide was found to delay gastric emptying. Co-administration of semaglutide at a dose of 1 mg decreased AUC0–60 min and Cmax of paracetamol by 27 % and 23 %, respectively. The total exposure of paracetamol (AUC0–5 h) did not change. At co-administration of semaglutide and paracetamol, no dose adjustment of the latter is required.
Oral contraceptives
Semaglutide is not expected to reduce the effects of oral hormonal contraceptives. When co-administered with a combined oral hormonal contraceptives (0.03 mg ethinyl estradiol/0.15 mg levonorgestrel) and semaglutide, the latter did not have a clinically meaningful effect on the total exposure of ethinyl estradiol and levonorgestrel. Ethinyl estradiol exposure was not affected; there was a 20 % increase in levonorgestrel exposure at steady state. Cmax did not change for any of the components.
Atorvastatin
Semaglutide did not change the systemic exposure of atorvastatin after a single dose of atorvastatin (40 mg). The Cmax of atorvastatin decreased by 38 %. This change was considered clinically insignificant.
Digoxin
Semaglutide did not change the systemic exposure or Cmax of digoxin after a single dose of digoxin (0.5 mg).
Metformin
Semaglutide did not change the systemic exposure or Cmax of metformin after administration of metformin at a dose of 500 mg twice daily for 3.5 days.
Warfarin
Semaglutide did not change the systemic exposure or Cmax of R- and S-warfarin after a single dose of warfarin (25 mg). Based on the determination of international normalised ratio (INR), no clinically significant changes in warfarin PK effects were observed.
Incompatibility
Semavic® should not be mixed with other drug products, including infusion solutions. Substances added to Semavic® may cause semaglutide degradation.
Special warnings
The use of Semavic® is not recommended in patients with type 1 diabetes or for treatment of diabetic ketoacidosis. Semavic® is not a substitute for insulin.
GI reactions
The use of GLP-1R agonists may cause gastrointestinal ARs. This should be considered when treating patients with impaired renal function as nausea, vomiting, and diarrhea, may cause dehydration which could cause a deterioration of renal function.
Acute pancreatitis
Acute pancreatitis was observed with the use of GLP-1RA. Patients should be informed of the specific symptoms of acute pancreatitis. If pancreatitis is suspected, Semavic® treatment should be discontinued; if confirmed, Semavic® treatment should not be restarted. Caution should be taken in patients with a history of pancreatitis. With no other signs and symptoms of acute pancreatitis, increased pancreatic enzyme activity does not predict the development of acute pancreatitis.
Hypoglycaemia
Patients receiving Semavic® in combination with sulphonylurea derivative or insulin may have an increased risk of hypoglycaemia. The risk of hypoglycaemia may be lowered by a reduction of a dose of sulphonylurea derivative or insulin when initiating treatment with Semavic®.
Diabetic retinopathy
An increased risk of complications of diabetic retinopathy was observed in patients with diabetic retinopathy receiving therapy with insulin and semaglutide (see section “Side effect”). Semaglutide should be used with caution in patients with diabetic retinopathy receiving therapy with insulin. Such patients should be closely monitored and treated per clinical guidelines. Rapid improvement in glucose control was associated with temporary worsening of diabetic retinopathy, however, other reasons cannot be excluded as well.
Cardiac failure
There is no therapeutic experience of the use of Semavic® in patients with congestive heart failure class IV according to New York Heart Association (NYHA). For these patients, the medicinal product is contraindicated.
Thyroid disorders
Cases of medullary thyroid cancer (MTC) have been reported during post-marketing use of another GLP-1 analogue, liraglutide. The available data are insufficient to establish or exclude a causal relationship between MTC and the use of GLP-1 analogues. Patients should be advised about the risk of MTC and thyroid tumour symptoms (lump in the neck, dysphagia, shortness of breath, persistent hoarseness). A significant increase in the plasma calcitonin concentration may indicate MTC (in patients with MTC, the plasma calcitonin concentrations are usually >50 ng/L). With the increase in the plasma calcitonin concentrations, patients should be further examined. Patients with thyroid nodules identified during a physical examination or thyroid ultrasound should also be further examined. Semaglutide is contraindicated in patients with a personal or family history of MTC or type 2 MEN.
Non-clinical safety data
Non-clinical data from safety pharmacology, repeated dose toxicity and genotoxicity studies did not show any danger for humans. In 2-year carcinogenicity studies in rats and mice, semaglutide at clinically significant concentrations caused medullary thyroid cancer without fatal outcomes. Non-fatal medullary thyroid cancer observed in rats is typical of the GLP-1 analogue group. The risk is considered to be low for humans but cannot be completely excluded.
Fertility
The effect of semaglutide on human fertility is unknown. Semaglutide had no effect on male rat fertility. Among female rats, an increase in estrous cycle and a slight decrease in a number of ovulations were observed at doses that decreased female body weight.
Effects on ability to drive or operate machines
Semavic® has no or negligible effects on the ability to drive or operate machines. Patients should be warned to avoid hypoglycaemia when driving or operating machinery, especially when using Semavic® in combination with a sulfonylurea or insulin.
Instruction for Use
Semavic® pre-filled pen injector allows for dosing 0.25 mg, 0.5 mg, and 1 mg. One pen injector contains 3 mL of the solution. The Semavic® package contains disposable needles. The patient should be advised to discard the injection needle after each dosing according to local disposal regulations. Semavic® pen injector must not be shared with other people. Do not inject Semavic® if it looks other than clear, colorless or almost colorless solution. Do not inject Semavic® that has been frozen. Inject Semavic® only with the needles up to 8 mm long. The pen injector is to be used with disposable injection needles. After each injection, remove the needle and keep the Semavic® pen injector without a needle. It will help prevent needle blocking, contamination, infection, solution leakage, and injection of a wrong dose.
Presentation
Solution for subcutaneous injection, 1.34 mg/mL 3 mL in a colorless neutral glass cartridge with a rubber plunger sealed with a flip-off cap with a rubber disc. Each cartridge is installed in a plastic multi-dose disposable pen injector for multiple injections. A polypropylene film label is affixed to each pen injector’s body. 1 pre-filled multidose disposable pen injector for multiple injections and 4 disposable needles along with instruction for medical use and user manual are placed in a carton.
Shelf life
2 years. Do not use after the expiry date as labeled on a pen injector and package.
Storage conditions
Store at 2 °C to 8 °C (in a refrigerator), but not close to a freezer compartment. Keep away from light. Do not freeze. Keep the pen injector with medicinal product that is in use or carried as a spare at a temperature below 30 °C or at 2 °C to 8 °C (in a refrigerator) for 6 weeks. Do not freeze. Having taken a dose, put the cap on the pen injector to protect it from light. Protect Semavic® from excessive heat and light. Keep out of the reach of children.
Marketing status
Prescription only.
Marketing authorisation holder
OOO “GEROPHARM”, 9, ul. Zvenigorodskaya, Saint Petersburg, 191119 Russian Federation. Telephone: (812) 703-79-75 (multi-line), fax: (812) 703-79-76
Manufacturer
OOO “GEROPHARM”, Russian Federation
Manufacturer
OOO “GEROPHARM”, Russian Federation
All steps of the manufacturing process:
1. Unit 1, bldg. 5, “Kvartal A” area, Obolensk work settlement, Serpukhov city district, Moscow region, 142279 Russia.
2. Unit 82, bldg. 4, “Kvartal A” area, Obolensk work settlement, Serpukhov city district, Moscow region, 142279 Russia.
Organisation accepting consumer complaints
OOO “GEROPHARM”, 4, bldg. 1, Yachevsky proezd, Pushkin, Saint Petersburg, 196608 Russian Federation. Telephone: +7 (812) 493-55-01 Fax: +7 (812) 493-55-69 Hotline: 8-800-333-4376 (toll-free in Russia) Website: www.geropharm.ru
Please forward information of any adverse reactions at inform@geropharm.ru; farmakonadzor@geropharm.com or at the contacts of OOO “GEROPHARM” stated above.
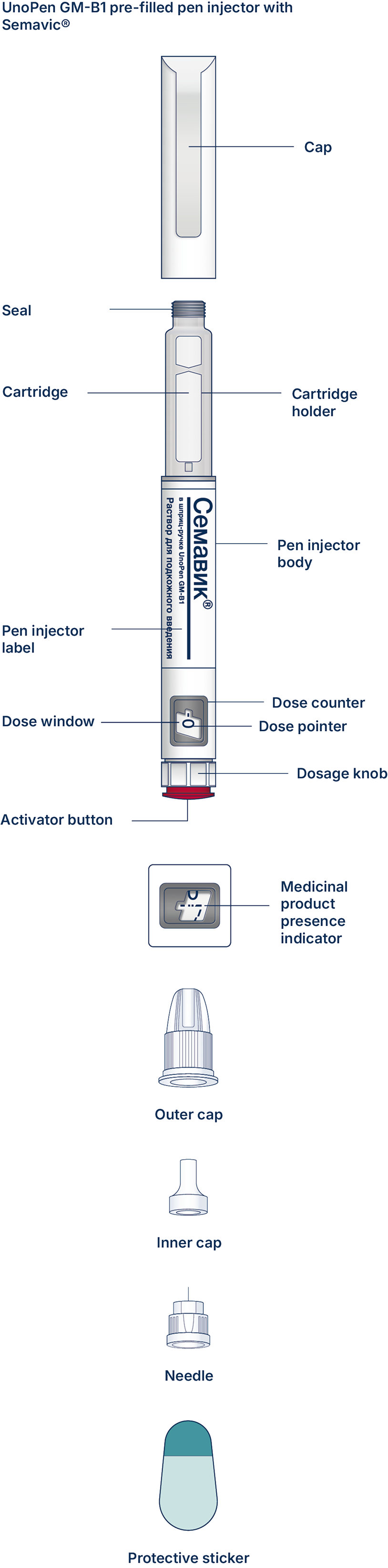
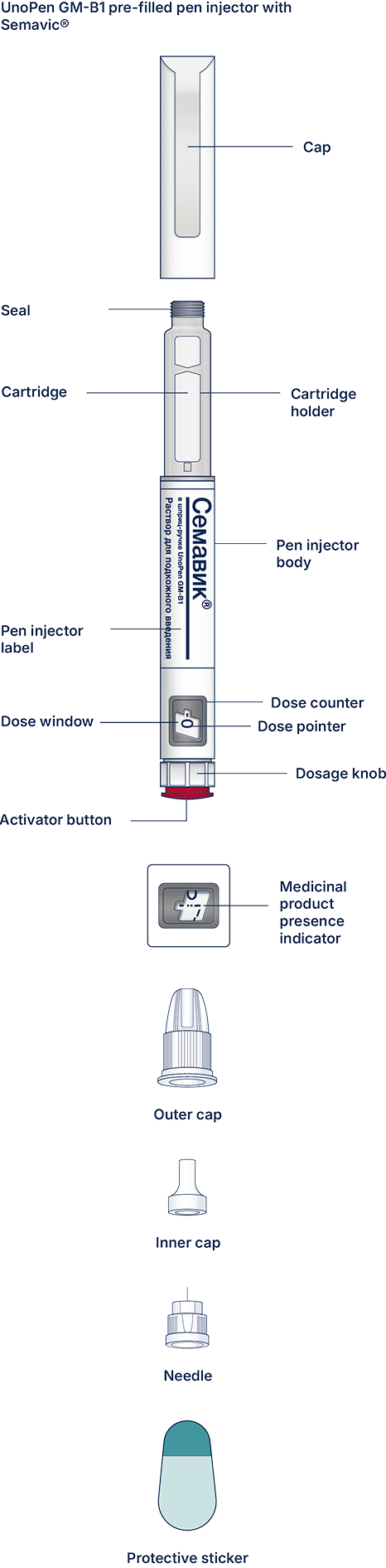
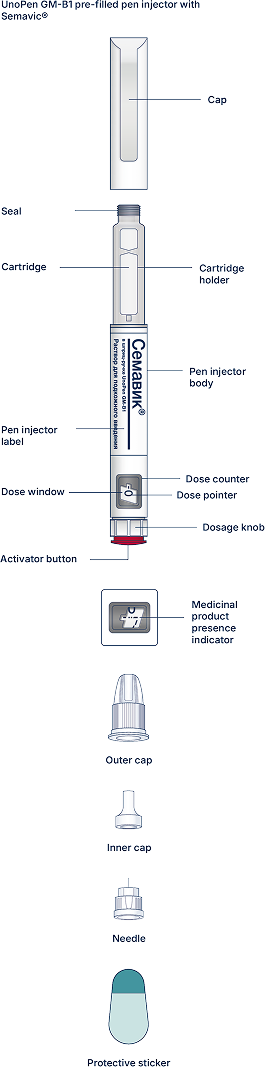
UnoPen GM-B1 pen injector is a pre-filled disposable pen injector for multiple injections (“pen injector”) with semaglutide, an antidiabetic drug, glucagon-like peptide-1 (GLP-1) analogue Semavic® (0.25 mg/dose, 0.5 mg/dose, 1.0 mg/dose), solution for subcutaneous injection in a pre-filled pen injector for delivery of the doses of 0.25 mg, 0.5 mg, and 1.0 mg. The pen injector is designed to titrate a dose of 0.25 mg and maintain a therapeutic dose of 0.5 mg and 1.0 mg. One pen injector contains 3 mL of the solution.
Semavic® can be injected using the pen injector in the following ways:
Option 1 - Dose titration
4 × 0.25 mg injections
4 × 0.5 mg injections
1 × 1.0 mg injection
Option 2 - Maintaining the therapeutic dose
4 × 1.0 mg injections
The pen injector is designed to be used with BD Micro-Fine™ Plus disposable injection needles. Each Semavic® package includes 4x WellFine 4 mm 32G (or Dexfine 4 mm 32G, or Verifine 4 mm 32G) needles.
Inject Semavic® only with the needles up to 8 mm long.
Make sure to attach a new needle before each injection. After an injection, store and carry the pen injector without a needle! It prevents needle blocking and contamination, infection, solution leakage, and injection of a wrong dose. Needles must be disposed in accordance with local requirements, rules and regulations for handling potentially infectious materials. One pen injector may used only by one person at a time. Make sure to never share your pen injector with anyone.
If Semavic® in the pen injector looks not like a colourless clear solution, then don’t use it.
Do not expose the pens to low or high temperatures (below +2 °C or above +30 °C). Do not put pen injectors in a freezer. Pen injectors must not be frozen!
Used pen injectors must be disposed and not be reused (don’t try to refill them).
Make sure to carry the pen injector in a special thermal case/bag (such as the original GEROPHARM case) when the temperature is too high or low.
Keep the pen injector and its needles out of the reach of everyone, especially children.
Do not try fixing the pen injector by yourself. If the pen injector is broken, contact the organization that accepts consumer claims as indicated in the instruction for medical use.
GEROPHARM helpline: 8 (800) 333-43-76.
Solution for subcutaneous injection in a pre-filled pen injector (disposable, multidose, for multiple injections)
Please read carefully this leaflet before using the Semavic® pre-filled pen injector.
Use the pen injector only after you learn how to use it under the guidance of a physician or a nurse.
At first, check the pen injector to make sure that it contains Semavic® 0.25 mg/dose, 0.5 mg/dose or 1 mg/dose. Then look at the figures below to get familiar with different components of the pen injector and needle.
If you have poor eyesight or serious vision problems, and cannot distinguish numbers on a dose counter, do not use the pen injector without help. Get help from a person with good eyesight who is trained to use the pre-filled pen injector of Semavic®.
This is a pre-filled pen injector. It contains 4 mg of semaglutide and allows you to select 0.25 mg, 0.5 mg or 1 mg. The pen injector is designed to be used with disposable needles up to a length of 8 mm.
The needles are included in a package.
Important information
Pay particular attention to the information marked with the symbols, this is very important for the safe use of a pen injector.
Discard a needle after every injection to ensure a comfortable injection and avoid needle blockage. If a needle is blocked, you cannot inject the medicinal product. Dispose of empty pen injectors with detached needles in accordance with local regulations or the recommendations given by your physician, nurse or pharmacist. Never try to put the inner cap back on the needle. You can prick yourself. Always immediately remove the needle from the pen injector after each injection. It helps prevent needle blocking and contamination, infection, solution leakage, and injection of a wrong dose.
Additional important information
Caring for your pen injector
Handle the pen injector carefully. Careless handling or misuse can lead to wrong dosing of the medicinal product which may result in high blood glucose or abdominal discomfort (nausea or vomiting).
Before the first injection, please read carefully the instructions for use of the UnoPen GM-B1 pre-filled disposable pen injector.
Please also consult your physician (specialist) on how to use the pen injector. Ask them to demonstrate the proper use of the pen injector. The first injection of the medicinal product must be carried out under the supervision of a physician or nurse.
Carefully read the label of the pen injector and make sure this is the medicinal product and the dose that your physician prescribed to you. Also check the medicinal product expiry date. Then look at the pictures below to get familiar with the characteristics and components of the pen injector.
Before getting started with the pen injector, check it carefully for visible mechanical damage or leakage (this may happen when the cartridge is not properly sealed). Never use the pen injector if you are not sure that it is functional and not damaged. Always check your pen injector before each injection.
Follow the user manual carefully: do not drop it and make sure to protect it from environmental factors (heat, direct sunlight, damage, physical damage, etc.). It the pen injector is damaged, replace it with another one immediately.
Visually impaired patients or patients with severe vision issues who cannot see the numbers on the dose counter must use the pen injector only under the supervision of medical personnel, relatives or any person with good vision who has been trained to inject medicinal products using pre-filled pen injectors.
Caregivers must handle used needles with extreme caution to prevent accidental injections and cross-infections.
The UnoPen GM-B1 pre-filled pen injector with Semavic®
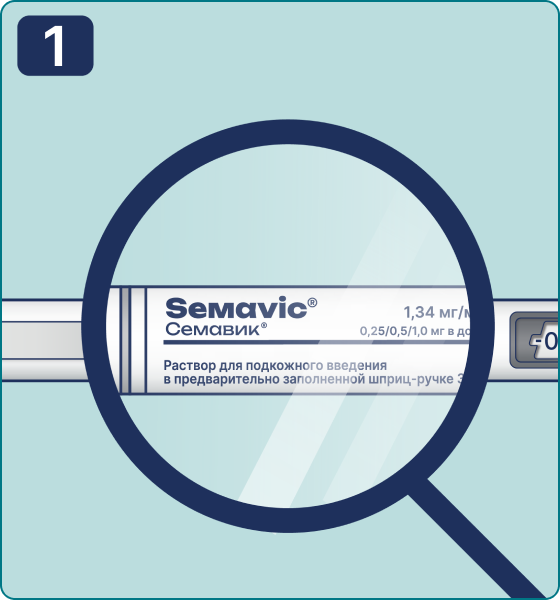
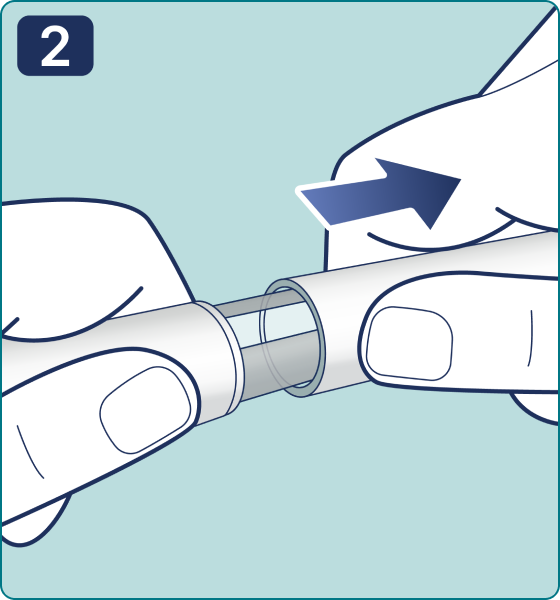
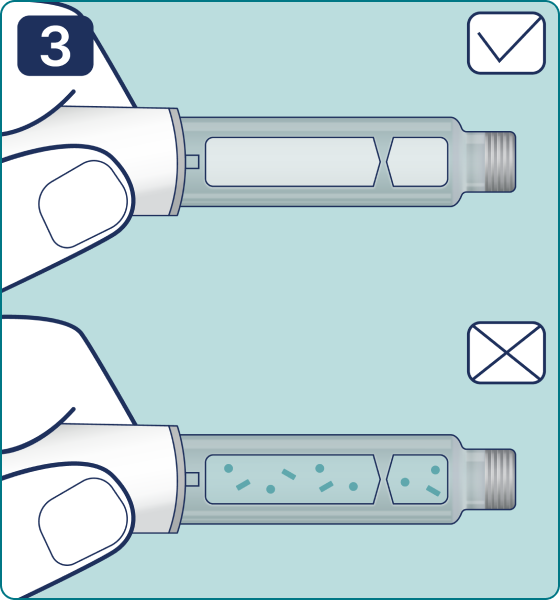
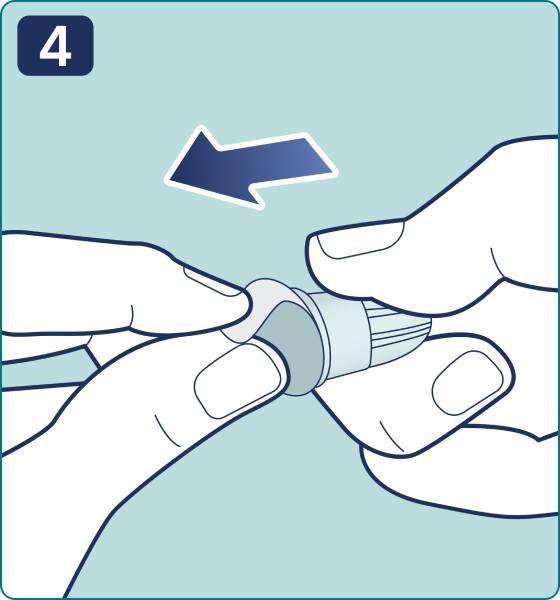
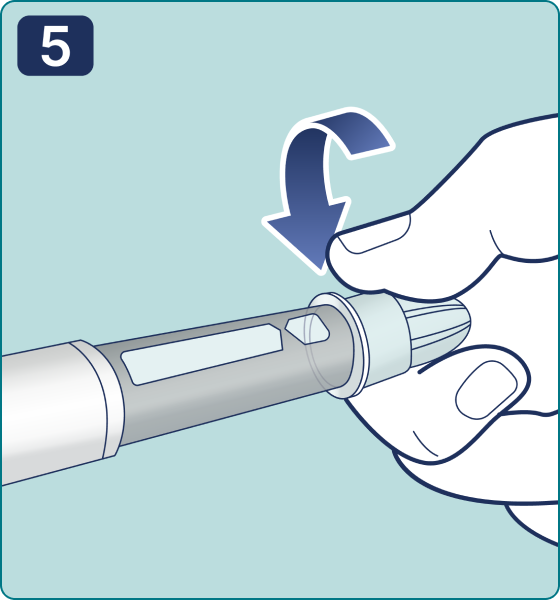
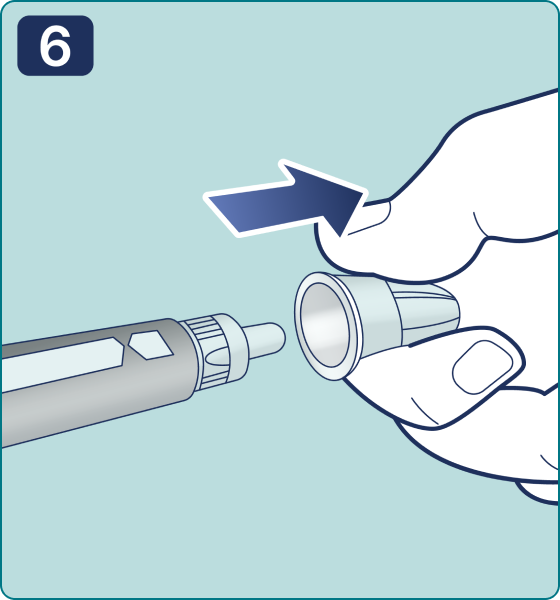
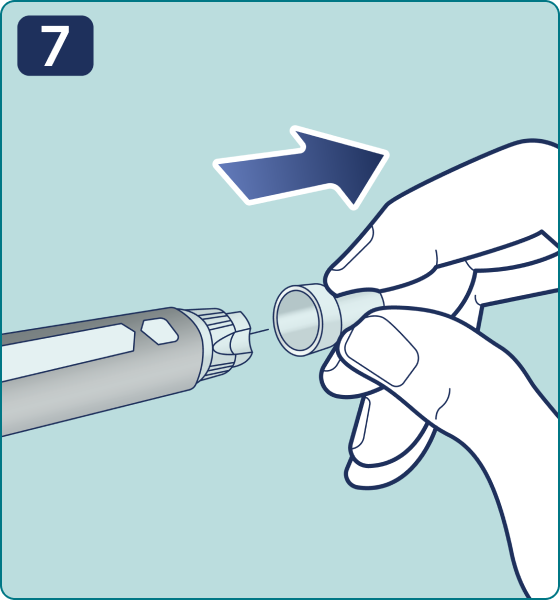
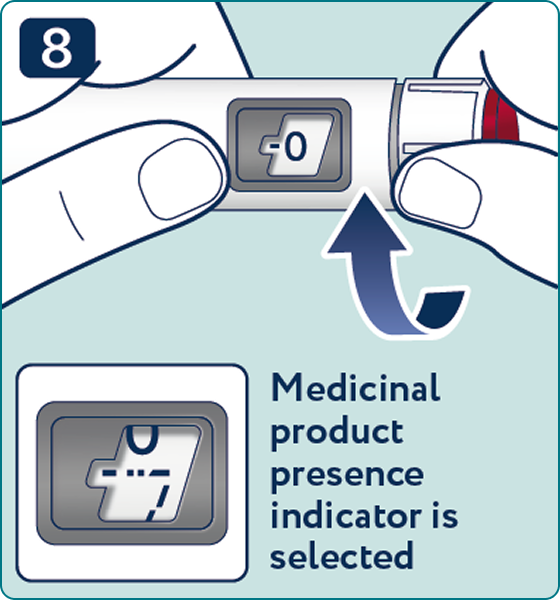
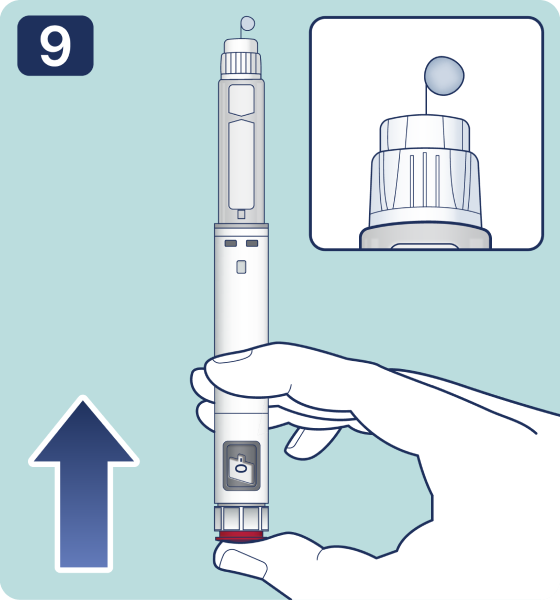
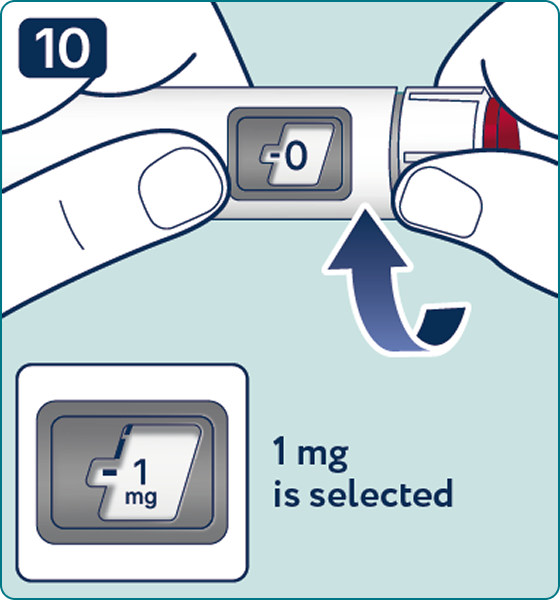
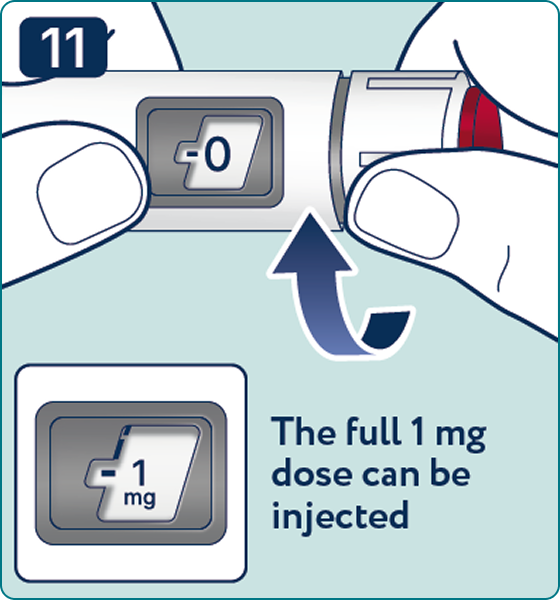
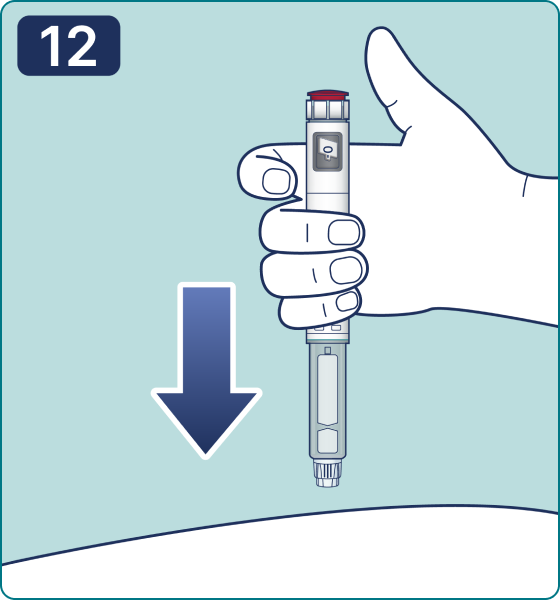
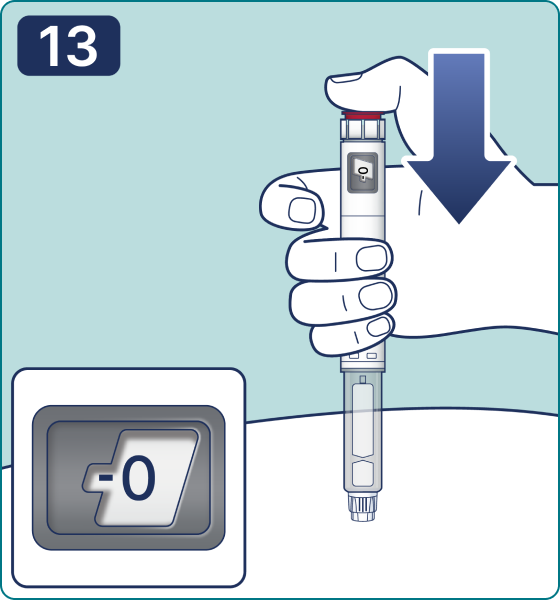
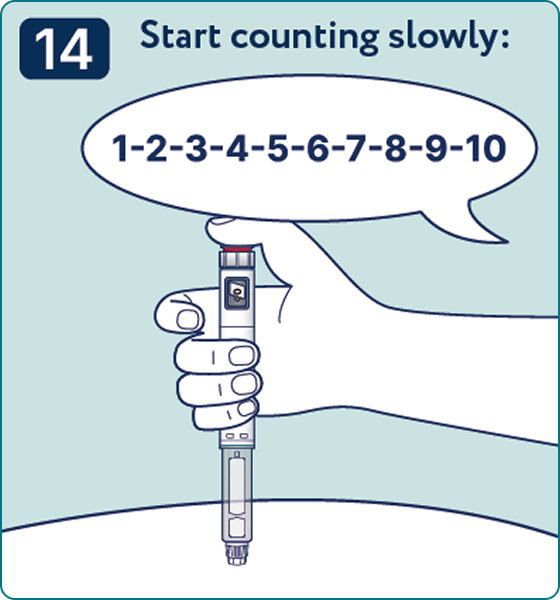
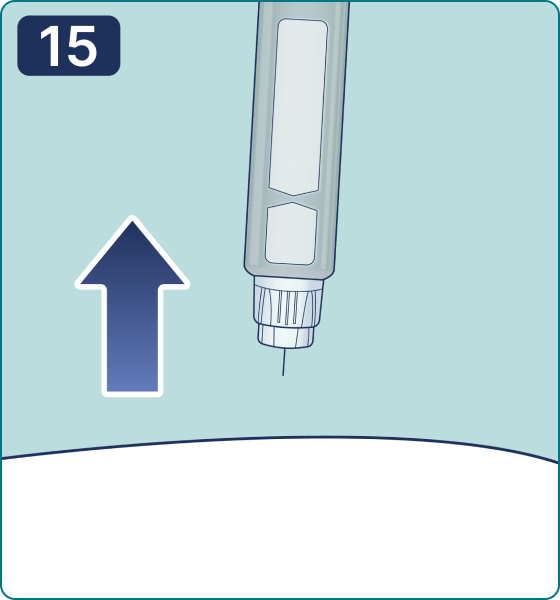
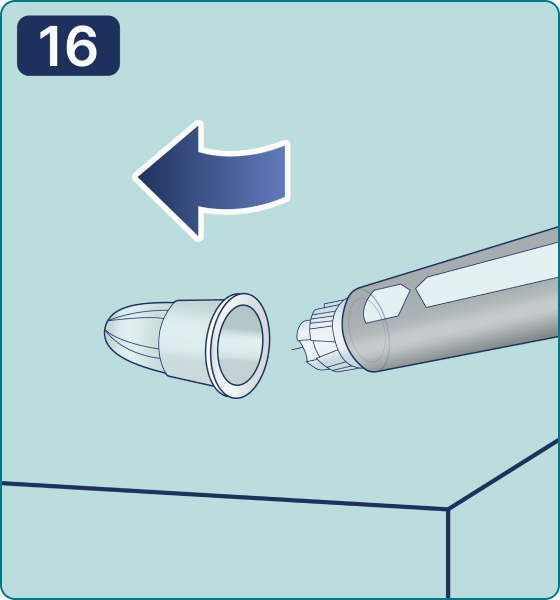
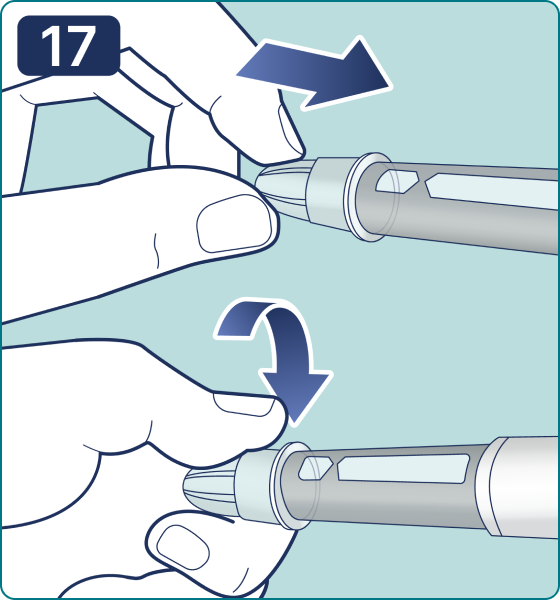
Rotate the dose selector until the « » solution pass indicator is aligned with the dose pointer. You will hear 2 (two) clicks while turning the selector to the check indicator.
» solution pass indicator is aligned with the dose pointer. You will hear 2 (two) clicks while turning the selector to the check indicator.
Note: If the dosage selector has skipped the required symbol, turn it in the opposite direction to adjust the position of the check symbol. If the pen is already in use, start using it from the "Setting the dose" item.